EN
New research co-led by Dr. Runze Yu from HPSTAR introduces an entropy-driven defect-engineering strategy that enables precise regulation of coupled cation–anion vacancies and dynamic surface reconstruction in high-entropy electrocatalysts. The results are published in Advanced Functional Materials.
Electrocatalytic water splitting is central to sustainable hydrogen production, yet its efficiency is fundamentally limited by the sluggish kinetics of the oxygen evolution reaction (OER), a complex multielectron process involving high energy barriers. In recent years, high-entropy materials (HEMs) have emerged as a promising platform due to their compositional diversity, tunable electronic structures, and synergistic multi-element interactions. These features provide unprecedented opportunities to optimize adsorption energetics and catalytic pathways. However, despite extensive efforts in defect engineering, a key challenge remains: the lack of precise control over coupled multisite defects, particularly the interplay between cation and anion vacancies, and their dynamic evolution under operating conditions. Conventional strategies often rely on post-synthetic treatments or in situ defect formation, which suffer from poor controllability and limited structural stability, thereby hindering the establishment of clear structure–activity relationships and rational catalyst design.
In this work, the researchers develop an entropy-stabilized cation–anion vacancy coupling strategy using high-entropy metal phosphorus trisulfides as a model system. By tuning cation stoichiometry, they achieve semi-quantitative control of coupled vacancies within a single-phase lattice, leading to asymmetric coordination environments, lattice distortion, and optimized electronic structures. Operando spectroscopy reveals that such vacancy coupling promotes earlier and more stable surface reconstruction into active oxyhydroxide species while suppressing excessive degradation. As a result, the optimized (MnFeCoNiZn)0.7PS3−δ catalyst exhibits excellent OER performance, delivering a low overpotential of 212 mV at 10 mA cm-2, a small Tafel slope of 28.6 mV dec-1, and outstanding durability over 800 h at high current densities, together with strong potential in practical alkaline electrolyzers. This work establishes entropy-enabled vacancy coupling as a general design principle for controlling defect chemistry and dynamic reconstruction, offering new insights into the rational design of high-performance electrocatalysts for sustainable energy conversion.
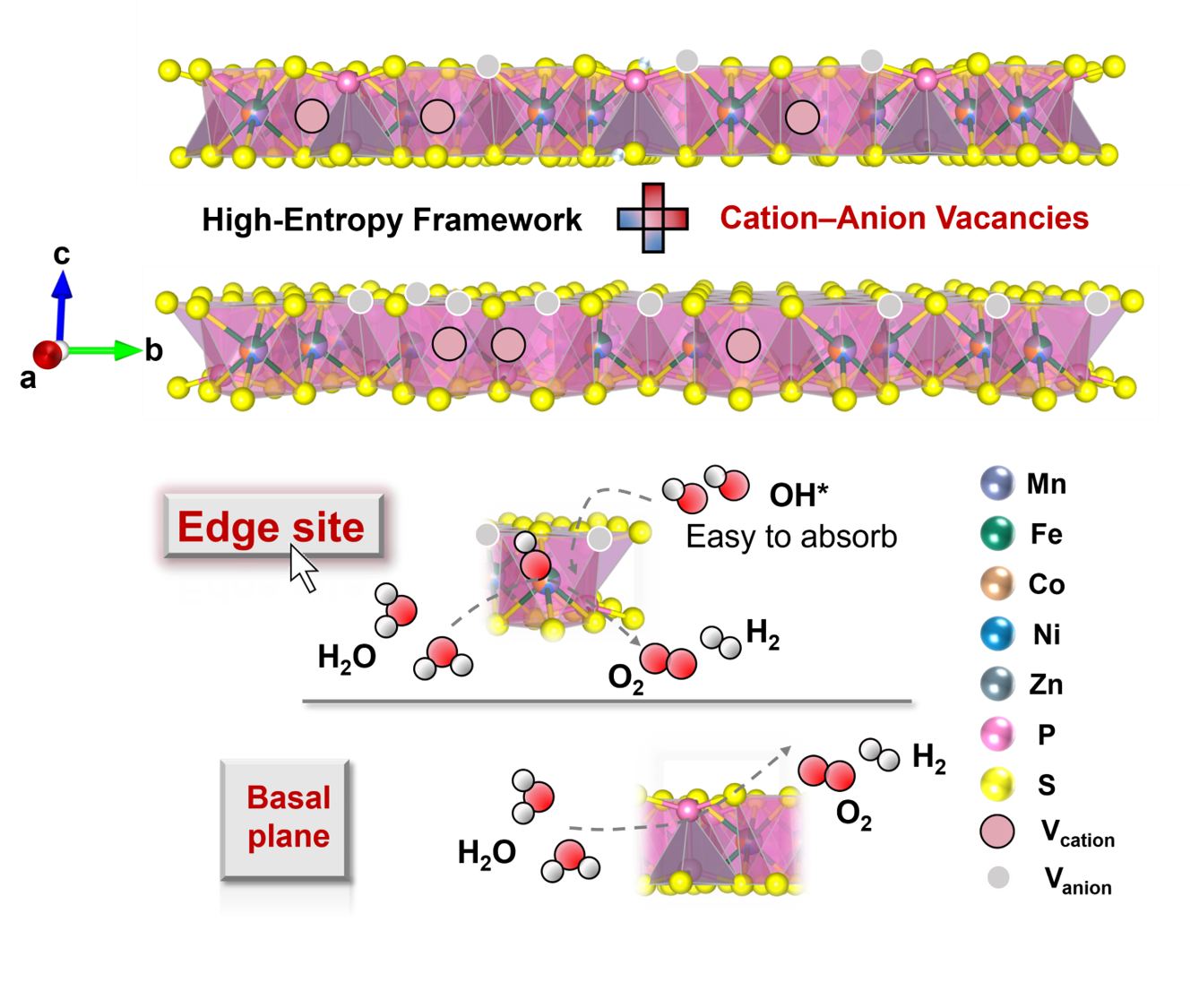
Caption: Entropy-stabilized cation-anion vacancies coupling in high-entropy metal phosphorus trisulfides (MPS3) induces lattice distortion and asymmetric coordination, enabling precise electronic modulation and synergistic Co-edge/P-basal active motifs.
开发高效且稳定的析氧反应(OER)电催化剂是实现高效电解水制氢的关键科学问题。然而,OER涉及复杂多电子转移过程,动力学缓慢,严重制约能量转化效率。近年来,高熵材料因其多组分协同效应和可调电子结构,成为电催化研究的重要方向。近期,北京高压科学研究中心(HPSTAR)于润泽研究员团队在该领域取得重要进展。该研究以高熵金属磷硫化物(MPS3)为模型体系,提出熵稳定的阳离子–阴离子空位耦合调控策略。通过调节金属位点化学计量比,实现了金属与硫空位的协同构筑,形成具有非对称配位和晶格畸变的高熵结构。该耦合缺陷有效诱导电荷重分布,优化关键含氧中间体吸附,从而提升催化本征活性。原位拉曼结果表明,该策略可促进反应初期更可控的表面重构,加速活性氧羟化物生成,同时抑制过度氧化与结构退化。理论计算进一步揭示,过渡金属位点主导反应动力学,而磷位点提供协同路径,共同降低反应能垒。得益于上述结构与机制优势,优化后的(MnFeCoNiZn)0.7PS3−δ催化剂表现出优异的OER性能,在10 mA cm-2下仅需212 mV过电位,Tafel斜率低至28.6 mV dec-1,并可在200 mA cm-2高电流密度下稳定运行超过800小时。在实际碱性电解槽中,该体系在1 A cm-2电流密度下仅需1.8 V电压,展现出良好的应用潜力。该研究建立了“高熵调控-空位耦合-动态重构-催化性能”之间的内在关联,为复杂多元体系中缺陷工程与电催化性能调控提供了新的设计范式,对高效电解水催化材料的开发具有重要指导意义。相关结果于近期以“Entropy-Stabilized Cation–Anion Vacancies Coupling in High-Entropy Two-Dimensional Crystals for Efficient Oxygen Evolution”为题发表于Advanced Functional Materials。

自由 Freedom
合作 Collaboration
顶尖 Excellence